利巴韦林
利巴韦林又名病毒唑、三氮唑核苷、尼斯可等,是广谱强效的抗病毒药物,目前广泛应用于病毒性疾病的防治。常用剂型有注射剂、片剂、口服液、气雾剂等。利巴韦林副作用少,不良反应发生率低,根据其药理作用,应用时要注意大剂量长期使用本药可引起白细胞减少、贫血、血清转氨酶和胆红素升高。应避免盲目、超量用药。大剂量使用本药所引起的胆红素增高和血红蛋白下降,可对临床检验和诊断造成干扰。
目录 |
基本信息
【通用名】 利巴韦林
【化学名】 1-β-D-呋喃核糖基-1H-1,2,4,-三氮唑-3-羧酰胺
【拼音名】 LIBAWEILIN
【英文名】 RIBAVIRIN
【标示】CAS编码:36791-04-5
ATC编码:J05AB04
PubChem:5064
DrugBank:APRD00081
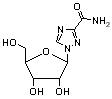
【结构式】见右图
【分子式】 C8H12N4O5
【分子量】 244.21
【别名】病毒唑(Bingduzuo) 利巴韦林(Ribavirin),三氮唑核苷(Ribavirin),威乐星,Virazde,RBV。
适应症
①幼儿呼吸道合胞病毒肺炎。②甲型、乙型流感和副流感病毒感染。③流行性出血热。④单纯疱疹。⑤麻疹、腮腺炎、水痘、带状疱疹等。
不良反应
本品不良反应较少,且多为可逆性。①贫血、白细胞减少。②皮疹、腹泻、胃肠道出血。③血清胆红素升高。
注意事项
1. 孕妇禁用,对本品过敏者禁用。连用不超过7日。
2. 有严重贫血、肝功能异常者慎用。
3. 对诊断的干扰:口服本品后引起血胆红素增高者可达25%。大剂量可引起血红蛋白下降。
4. 尽早用药:呼吸道合胞病毒性肺炎病初3日内给药一般有效本品不宜用于未经实验室确诊为呼吸道合胞病毒感染的患者。
5. 长期或大量用药对肝功能、血象有不良反应。
制剂
1.片剂:100mg。2.口服液:150mg/5ml、300mg/10ml。3.针剂:100mg/1ml。4.眼药水:8mg/8ml。5.滴鼻液:50mg/10ml。
用法用量
1.口服
成人每次mg~200mg,老人每次mg~150mg,每日3次,小儿每日mg/kg~15mg/kg,分3次服。
2.肌肉注射或静脉滴注
成人及小儿每日mg/kg~15mg/kg,老人每日mg/kg,分2次肌肉注射或静脉滴注。
3.滴眼、滴鼻
每1h~2h1次,每次滴~2滴。
4.气雾吸入
可将本品50mg~100mg的注射剂加入10ml~20ml生理盐水中,超声雾化吸入,每日2次。
药物毒理
药理作用
利巴韦林为合成的核苷类抗病毒药。体外细胞培养试验表明,利巴韦林对呼吸道合胞病毒(RSV)具有选性的抑制作用。利巴韦林的作用机理尚不清楚,但是其体外抗病毒活性可被鸟嘌呤核苷和黄嘌呤核苷逆转的结果提示,利巴韦林可能作为这些细胞的代谢类似物而起作用。
毒理研究
重复给药毒性:小鼠、大鼠和猴在经口给予利巴韦林剂量分别为30、36和120mg/kg,给药时间为4周或更长时,可引起心脏损伤。
遗传毒性:利巴韦林浓度分别为0.015和0.03-5.0mg/ml,在无代谢活化物条件下,可增加小鼠Balb/c3T3(成纤维细胞)和L5178Y(淋巴瘤)的细胞转化和突变。浓度范围为3.75-10.0mg/ml在加入代谢活化物条件下,对L5178Y细胞突变率有一事实上的增加(3-4倍)。小鼠微核试验结果提示,静脉注射利巴韦林剂量范围为20-200mg/kg时,具有诱裂作用。在显性致死试验中,大鼠腹腔注射利巴韦林剂量范围为50-200mg/kg,连续5天,未见有致突变作用。
生殖毒性:雄性小鼠给予剂量范围在35-150mg/kg时,可导致明显的生精管萎缩,精子浓度降低和形态异常的精子数量增加。停药后3-6个月,生精能力部分恢复。其它几项毒性试验也提示,成年大鼠经口给予利巴韦林剂量低至16mg/kg时,可引起睾丸操作(生精管萎缩),未进行更低剂量的研究。尚未对雄性动物的生死能力进行研究。不同种属的动物研究已证实利巴韦林有明显的致畸和/或杀胚和1.0mg/kg,结果均已证实有致畸作用。畸形主要发生在颅骨、腭、眼、四肢、颌骨、骨骼和胃肠道,其发生率和严重程序随剂量的递增而增加。胎儿和子代的存活率降低。利巴韦林引起家兔和大鼠胚胎致死的剂量为1mg/kg,其无致畸作用剂量分别为0.1和0.3mg/kg(根据表面积推算,分别相当于人等效剂量0.015和0.04mg/kg)。
致癌性:大鼠经掺食给予利巴韦林剂量为16-200mg/kg长期研究结果提示,利巴韦林可能诱发良性乳房、胰管、垂体和肾上腺瘤。小鼠和大鼠的18-24个月的初步致癌试验并非最终结果,但这试验证实,给予利巴韦林剂量分别为20-75和10-40 mg/kg,小鼠和大鼠分别出现的血管损伤和视黄醛还原酶变性与利巴韦林长期给药有关。
【药代动力学】
国内人体生物利用度研究资料表明,利巴韦林颗粒口服后吸收迅速,在60-90分钟内血药浓度可达到峰值。利巴韦林进入体内后,经磷酸化生成具有活性的代谢产物—利巴韦林单磷酸。消除半衰期约为24小时。利巴韦林能滞留于红细胞内。主要由肾脏排泄,仅有少量随粪便排出。
据Physician’s Desk Reference(54版)介绍,慢性肝炎患者服用利巴韦林单剂量和多剂量的药动性质概括在表1中。口服后利巴韦林吸收迅速而完全。然而由于首过效应,绝对生物利用度平均值为64%(44%)。在单次服用200~1200mg剂量范围内,利巴韦林的剂量与AUC0-t(从0时间到最后测试点间的AUC)之间存在线性关系。但剂量与Cmax之间的关系呈曲线性的,单剂量在400~600mg以上时趋近于渐近线。
多次口服后,可以观察到在血浆中有6倍的利巴韦林蓄积(以AUC12hr为基础)。连续口服600mg,每日两次,大约四周可以达到稳态,稳态血浆平均浓度为2200(37%)ng/ml,停药后测量的平均半衰期为298(30%)小时,这表明本品可能存在从非血浆部分缓慢消除。
食物对利巴韦林吸收的影响:在单剂量药物研究中,当利巴韦林与高脂肪餐(841千卡热量,53.8g脂肪,31.6蛋白质和57.4g糖类)一同食用时,AUCtf和Cmax增加70%。尚没有足够的数据来证实这些结果的临床相关性。临床药效研究时未进行关于食物消耗方面的说明。(见用法用量)
抗酸剂对利巴韦林吸收的影响:服用利巴韦林的同时服用一种抗酸剂包含镁、铝和二甲硅油,会导致利巴韦林AUCtf平均值下降14%。单剂量研究结果临床相关性未知。[见表1]
表1慢性肝炎成年患者服用利巴韦林后的平均药代动力学参数(N=12)
| 参 数 | 利巴韦林剂量(变异系数) | |
| 单剂量600mg | 多剂量600mg每日两次 | |
| Tmax | 1.7(46)... | 3(60) |
| Cmaxng/ml | 782(37) | 3680(85) |
| T1/2(hr) | 43.6(47) | 298(30) |
| AUCtf..ng.h/ml | 13400(48) | 228000(25) |
| 表观分布容积(L) | 2825(9)+ | |
| 表观清除率(L/hr) | 38.2(40) | |
| 绝对生物利用度 | 64%(44)++ |
利巴韦林能进入红细胞内,并已被确认通过es—型核苷载体进入的。实质上这种类型载体存在于所有类型的细胞中,可以导致分布容积扩大。利巴韦林与血浆蛋白结合少。
利巴韦林有两种代谢途径:(i)一种是在有核细胞中可逆的磷酸化;(ii)另一种是包括脱核糖基化和胺水解产生一种三吡咯羧酸代谢物的代谢途径。利巴韦林及其三吡咯酰胺和三吡咯羧酸代谢物经肾排泄。口服600mg14C-利巴韦林后,在336小时内在尿及粪便中分别有61%和12%左右消除,其中未经转化的利巴韦林只占17%。
人及大鼠肝脏微粒体体外代谢研究结果表明:利巴韦林很少或几乎不通过细胞色素P450代谢,只有极少量潜在的酶—药物之间相互作用。
特殊人群
肾功能障碍:患有不同程度的肾功能障碍的HCV感染患者中服单剂量(400mg )的利巴韦林后,肌酐清除率值在10~30ml/min的患者较对照组(肌酐清除率>90ml/min)AUCtf值大了3倍,较肌酐清除率值在30~60ml/min的患者AUCtf值大了2倍,这均是因为清除率下降减少了药物消除。多次给药后利巴韦林的药代参数很难预测。血液透析不能有效清除利巴韦林。肌酐清除率<50ml/min的患者,不推荐使用利巴韦林(见注意事项)。
肝功能障碍:具有轻、中、重度肝功能障碍患者(按Chaild-pugh分类为A、B、C)分别口服单剂量(600mg)的利巴韦林后,与对照组相比平均AUCtf值没有明显的不同。然而平均Cmax值随肝功能障碍的严重而增大,患有严重肝功能障碍的患者比对照组的Cmax值大2倍。
老年患者:尚未对老年患者进行药动学研究。
性别:在对18个男性患者及18个女性患者进行的单剂量研究中,没有发现明显的性别药动学不同。利巴韦林为合成的核苷类抗病毒药。体外细胞培养试验表明,利巴韦林对呼吸道合胞病毒(RSV)具有选择性的抑制作用。利巴韦林的作用机理尚不清楚,但是其体外抗病毒活性可被鸟嘌呤核苷和黄嘌呤核苷逆转的结果提示,利巴韦林可能作为这些细胞的代谢类似物而起作用。
重复给药毒性:小鼠、大鼠和猴在经口给予利巴韦林剂量分别为30、36和120mg/kg,给药时间为4周或更长时,可引起心脏损伤。
遗传毒性:利巴韦林浓度分别为0.015和0.03-5.0mg/ml,在无代谢活化物条件下,可增加小鼠Balb/c3T3(成纤维细胞)和L5178Y(淋巴瘤)的细胞转化和突变。浓度范围为3.75-10.0mg/ml,在加入代谢活化物条件下,对L5178Y细胞突变率有一定的增加(3-4倍)。小鼠微核试验结果提示,静脉注射利巴韦林剂量范围为20-200mg/kg时,具有诱裂作用。在显性致死试验中,大鼠腹腔注射利巴韦林剂量范围为50-200mg/kg,连续5天,未见有致突变作用。
生殖毒性:雄性小鼠给予剂量范围在35-150mg/kg时,可导致明显的生精管萎缩,精子浓度降低和形态异常的精子数量增加。停药后3-6个月,生精能力部分恢复。其它几项毒性试验也提示,成年大鼠经口给予利巴韦林剂量低至16mg/kg时,可引起睾丸损伤(生精管萎缩),未进行更低剂量的研究。尚未对雌性动物的生殖能力进行研究。不同种属的动物研究已证实利巴韦林有明显的致畸和/或杀胚的潜在毒性。仓鼠单次经口给予本品剂量为2.5mg/kg或更大,家兔或大鼠的剂量分别为0.3和1.0mg/kg,结果均已证实有致畸作用。畸形主要发生在颅骨、腭、眼、四肢、颌骨、骨骼和胃肠道,其发生率和严重程度随剂量的递增而增加。胎儿和子代的存活率降低。利巴韦林引起家兔和大鼠胚胎致死的剂量为1mg/kg,其无致畸作用剂量分别为0.1和0.3mg/kg(根据表面积推算,分别相当于人等效剂量0.015和0.04mg/kg)。
致癌性:大鼠经掺食给予利巴韦林剂量为16-200mg/kg长期研究结果提示,利巴韦林可能诱发良性乳房、胰管、垂体和肾上腺瘤。小鼠和大鼠的18-24个月的初步致癌试验并非最终结果,但这些试验证实,给予利巴韦林剂量分别为20-75和10-40mg/kg,小鼠和大鼠分别出现的血管损伤和视黄醛还原酶变性与利巴韦林长期给药有关。
药代动力学
国内人体生物利用度研究资料表明,利巴韦林颗粒口服后吸收迅速,在60-90分钟内血药浓度可达到峰值。利巴韦林进入体内后,经磷酸化生成具有活性的代谢产物-利巴韦林单磷酸。消除半衰期约为24小时。利巴韦林能滞留于红细胞内。主要由肾脏排泄,仅有少量随粪便排出。
据Physician’s Desk Reference(54版 )介绍,慢性肝炎患者服用利巴韦林单剂量和多剂量的药动性质概括在表1中。口服后利巴韦林吸收迅速而完全。然而由于首过效应,绝对生物利用度平均值为64%(44)。在单次服用200~1200mg剂量范围内,利巴韦林的剂量与AUC0-t(从0时间到最后测试点间的AUC)之间存在线性关系。但剂量与Cmax之间的关系呈曲线性的,单剂量在400~600mg以上时趋近于渐近线。
多次口服后,可以观察到在血浆中有6倍的利巴韦林蓄积(以AUC12hr为基础)。连续口服600mg,每日两次,大约四周可以达到稳态,稳态血浆平均浓度为2200(37%)ng/ml,停药后测量的平均半衰期为298(30%)小时,这表明本品可能存在从非血浆部分缓慢消除。
食物对利巴韦林吸收的影响:在单剂量药物研究中,当利巴韦林与高脂肪餐(841千卡热量,53.8g脂肪,31.6g蛋白质和57.4g糖类)一同食用时,AUCtf和Cmax增加70%。尚没有足够的数据来证实这些结果的临床相关性。临床药效研究时未进行关于食物消耗方面的说明。(见用法与用量)
抗酸剂对利巴韦吸收的影响:服用利巴韦林的同时服用一种抗酸剂包含镁、铝和二甲硅油,会导致利巴韦林AUCtf平均值下降14%。单剂量研究结果临床相关性未知。[见表1]
表1慢性肝炎 成年患者服用利巴韦林后的平均药代动力学参数(N=12)
┌───────────┬─────────────────────────┐
│ 参 数 │ 利巴韦林剂量(变异系数) │
├───────────┼─────────────┬───────────┤
│ │单剂量600mg │多剂量600 mg每日两次 │
├───────────┼─────────────┼───────────┤
│Tmax(hr) │1.7(46) │3(60) │
├───────────┼─────────────┼───────────┤
│Cmax ng/ml │782(37) │3680(85) │
├───────────┼─────────────┼───────────┤
│T1/2(hr) │43.6(47) │298(30) │
├───────────┼─────────────┼───────────┤
│AUCtf(ng.h/ml) │13400(48) │228000(25) │
├───────────┼─────────────┼───────────┤
│表观分布容积(L) │2825(9) │ │
├───────────┼─────────────┼───────────┤
│表观清除率(L/hr) │38.2(40) │ │
├───────────┼─────────────┼───────────┤
│绝对生物利用度 │64%(44) │ │
└───────────┴─────────────┴───────────┘
利巴韦林能进入红细胞内,并已被确认通过es-型核苷载体进入的。实质上这种类型载体存在于所有类型的细胞中,可以导致分布容积扩大。利巴韦林与血浆蛋白结合少。
利巴韦林有两种代谢途径:(i)一种是在有核细胞中可逆的磷酸化;(ii)另一种是包括脱核糖基化和胺水解产生一种三吡咯羧酸代谢物的代谢途径。利巴韦林及其三吡咯酰胺和三吡咯羧酸代谢物经肾排泄。口服600mg14C-利巴韦林后,在336小时内在尿及粪便中分别有61%和12%左右消除,其中未经转化的利巴韦林只占17%。
人及大鼠肝脏微粒体体外代谢研究结果表明:利巴韦林很少或几乎不通过细胞色素P450代谢,只有极少量潜在的酶-药物之间相互作用。
特殊人群
肾功能障碍:患有不同程度的肾功能障碍的HCV感染患者口服单剂量(400mg)的利巴韦林后,肌酐清除率值在10~30ml/min的患者较对照组(肌酐清除率>90ml/min)AUCtf值大了3倍,较肌酐清除率值在30~60ml/min的患者AUCtf值大了2倍,这均是因为清除率下降减少了药物消除。多次给药后利巴韦林的药代参数很难预测。血液透析不能有效清除利巴韦林。肌酐清除率<50ml/min的患者,不推荐使用利巴韦林(见注意事项)。
肝功能障碍:具有轻、中、重度肝功能障碍患者(按Chaild-pugh分类为A、B、C)分别口服单剂量(600mg)的利巴韦林后,与对照组相比平均AUCtf值没有明显的不同。然而平均Cmax值随肝功能障碍的严重而增大,患有严重肝功能障碍的患者比对照组的Cmax值大2倍。
儿科患者:尚未对儿科患者进行详细药动学研究。
老年患者:尚未对老年患者进行药动学研究。
性别:在对18个男性患者及18个女性患者进行的单剂量研究中,没有发现明显的性别药动学不同。
参看
| ||||||||
| ||||||||
| ||||||||||||||||||||||||||||||||||